Correct retention times in database according to internal standards
Xiaotao Shen (https://www.shenxt.info/)
Si Wu
School of Medicine, Stanford UniversityCreated on 2020-03-28 and updated on 2021-07-03
Source:vignettes/rt_correction.Rmd
rt_correction.RmdRetention time correction
The metabolite retention time (RT) may shift in different batches. Therefore, if you spike internal standards into your standards and biological samples, you can correct the retention times in database using the correct_database_rt() function.
Here we first get the database using the demo data from demoData package.
library(demoData)
library(metID)
path <- system.file("database_construction", package = "demoData")
file.copy(
from = path,
to = ".",
overwrite = TRUE,
recursive = TRUE
)
#> [1] TRUE
new.path <- file.path("./database_construction")new.path <- file.path("./database_construction")
test.database <- construct_database(
path = new.path,
version = "0.0.1",
metabolite.info.name = "metabolite.info_RPLC.csv",
source = "Michael Snyder lab",
link = "http://snyderlab.stanford.edu/",
creater = "Xiaotao Shen",
email = "shenxt1990@163.com",
rt = TRUE,
mz.tol = 15,
rt.tol = 30,
threads = 5
)
#> Reading metabolite information...
#> Reading positive MS2 data...
#> Reading MS2 data...
#> Processing...
#>
|
| | 0%
|
|============== | 20%
|
|============================ | 40%
|
|========================================== | 60%
|
|======================================================== | 80%
|
|======================================================================| 100%
#>
#> OK
#> Reading negative MS2 data...
#> Reading MS2 data...
#> Processing...
#>
|
| | 0%
|
|============== | 20%
|
|============================ | 40%
|
|========================================== | 60%
|
|======================================================== | 80%
|
|======================================================================| 100%
#>
#> OK
#> Matching metabolites with MS2 spectra (positive)...
#> OK
#> Matching metabolites with MS2 spectra (negative)...
#> OK
#> All done!Data preparation
First, please prepare two internal standard (IS) tables for the database and biological samples. The format of the IS table is shown in the figure below:
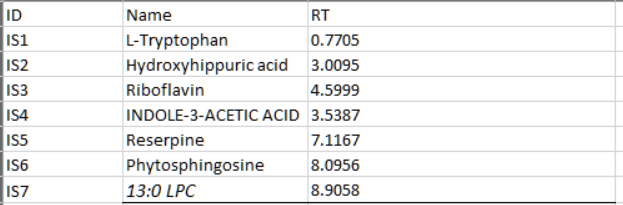 The IS table for the database should be named as “database.is.table.xlsx” and the IS table for experiment should be named as “experiment.is.table.xlsx”.
The IS table for the database should be named as “database.is.table.xlsx” and the IS table for experiment should be named as “experiment.is.table.xlsx”.
Run correct_database_rt() function
test.database2 <-
correct_database_rt(
experiment.is.table = "experiment.is.table.xlsx",
database.is.table = "database.is.table.xlsx",
database = test.database,
path = new.path
)The database should be the database (databaseClass object) for which you want to correct RTs.
Note:
test.database2is only a demo database (metIdentifyClass object). We will not use it for the next metabolite identification step.