Brief introduction of lipidflow
Xiaotao Shen PhD (https://www.shenxt.info/)
Chuchu wang PhD
School of Medicine, Stanford UniversityCreated on 2021-02-09 and updated on 2021-03-03
Source:vignettes/lipidflow.Rmd
lipidflow.RmdIntroduction & Installation
Installation
You can install lipidflow from Github.
if(!require(devtools)){
install.packages("devtools")
}
devtools::install_github("jaspershen/lipidflow")lipidflow is a part of tidymass, so you can also install it by tidymass.
Workflow of lipidflow
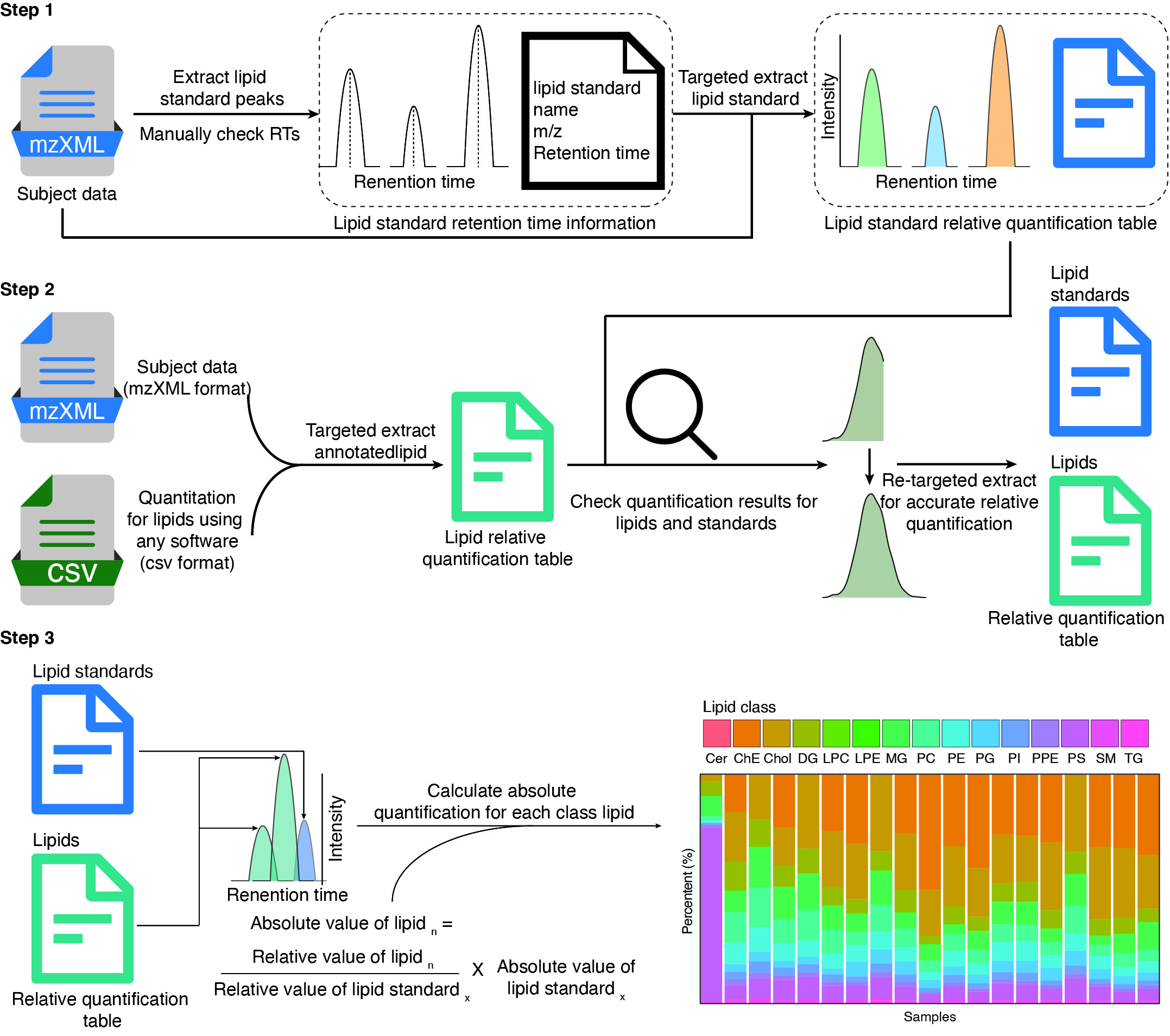
Step1: Get the retention time of internal standards
The first step is to extract the extracted ion chromatogram (EIC) of all internal standards (IS) in samples and then get the retention times (RTs), adduct and m/z of all the internal standards. Internal standards are used for absolute quantification of lipids.
Step 2: Get relative quantification data
In this step, we will get the relative quantification data of internal standards and lipids by targeted extracting from raw data (mzXML format). The lipid annotation table can be from other tools, here we recommend lipidSearch from ThermoFisher.
Step 3: Get absolute quantification data
In this step, we will get the absolute quantification data for all the lipids. The absolute quantification value for each lipid is calculated as:
\[ AI_i = \frac{RI_{i}AI_j}{RI_j} \]
Here, \(AI_i\) is the absolute intensity of \(i^{th}\) lipid. \(RI_{i}\) is the relative intensity of \(i^{th}\) lipid. \(AI_i\) is the absolute intensity of \(j^{th}\) internal standard. \(RI_i\) is the absolute intensity of \(j^{th}\) internal standard.
So for each lipid, we need a corresponding internal standard for absolute quantification. In our demo data, for each class of lipids, we usually use one or two internal standard for absolute quantification.